










INTRODUCCIÓN
La importancia de adherirse a un estilo de vida activo y saludable está bien documentada. En los adultos, la investigación ha demostrado la importancia de la actividad física en la prevención de enfermedades cardiovasculares, el síndrome metabólico, la diabetes tipo 2 y la obesidad junto con la promoción de la longevidad1-4. En los niños y adolescentes, una revisión reciente también indica que la práctica de alguna actividad física está asociada con una serie de efectos positivos en la salud5.
A medida que se han ido oficializando las actividades deportivas, ya sea con la participación en actividades reglamentadas o con más competiciones, un grupo más grande de deportistas han hecho el salto hacia una dedicación mayor a la competición amateur, en la que los mismos deportistas buscan la mejora constante en su capacidad de practicar deporte. No es extraño pues entrever que los deportistas busquen mejorar su rendimiento mediante entrenamientos más rigurosos y una nutrición más adecuada. A medida que se avanza en los conocimientos moleculares de la capacidad física, incluyendo los factores genéticos, hay más interés en esta tecnología, para predecir desde el tipo de ejercicio más conveniente hasta el riesgo de padecer una lesión muscular.
Pero dentro de este contexto de avances genéticos en el deporte sobresale la información referente a la muerte súbita en los deportistas, causada por enfermedades genéticas hereditarias. A pesar de que son poco frecuentes, la muerte súbita del atleta tiene un impacto mediático instantáneo que crea una alarma social importante.
Las ciencias de la actividad física y del deporte son un ámbito de investigación que tampoco puede escapar de los cambios que está experimentando la biología molecular y, en concreto, el campo de la genómica y de la genética humana. En este sentido, sea con el objetivo de estudiar el impacto de un programa de entrenamiento dirigido al rendimiento, analizar los efectos de la actividad física o evaluar el riesgo de padecer una enfermedad hereditaria asociada a la muerte súbita en los deportistas, la información del genoma humano pasa a ser un elemento importante en las diversas investigaciones.
GENÉTICA
Son muchos los acontecimientos históricos relevantes que se han sucedido para llegar a confeccionar el mapa genético humano. Desde la publicación del Origen de las especies (1859) de Charles Darwin, pasando por los experimentos con guisantes (1865) de Gregor Mendel hasta el lanzamiento del Proyecto Genoma Humano (1990), la elaboración detallada del mapa genético humano (1995) y la finalización de la secuencia del genoma humano (2003), los estudios de biología molecular han vivido los últimos 25 años una importante revolución y en la actualidad su influencia es muy relevante en el campo de las ciencias6.
Todo organismo vivo tiene su propia información genética contenida en la molécula de ADN (ácido desoxirribonucleico). En ella se encuentran las unidades de herencia, los genes. Los humanos tenemos 30.000 genes distribuidos en 23 pares de cromosomas localizados en el núcleo de les células (22 autosómicos y 1 par sexual) y un único cromosoma mitocondrial. Cada par de cromosomas (homólogo) tiene los mismos genes -de los que tenemos dos copias, denominadas alelos-. Cada gen contiene la información necesaria para la síntesis de una proteína, la unidad funcional del organismo, ya que el buen funcionamiento de éste se basa en la síntesis perfecta de todas las proteínas necesarias.
La molécula de ADN está formada por 4 tipos diferentes de nucleótidos, repetidos millones de veces. La mayoría del ADN no codifica por proteínas, y los fragmentos que lo hacen son los denominados genes. En la porción codificadora del gen cada grupo de tres nucleótidos codifica por un determinado aminoácido. Éste es un proceso en cadena; los primeros tres nucleótidos codifican el primer aminoácido y los tres siguientes el segundo, y así sucesivamente. Esta síntesis viene determinada por el orden que sigue la secuencia de nucleótidos en el ADN. La acumulación progresiva de todos los aminoácidos del gen da lugar a la creación de la proteína. En el genoma humano se ha descrito que al menos el 1,5% de éste contiene secuencias que codifican por proteínas7. El resto es material que puede ayudar al funcionamiento correcto, pero su función aún no está claramente establecida.
En ocasiones se puede producir la inserción, la deleción o el cambio en el orden de los nucleótidos. Son defectos genéticos, denominados mutaciones, que pueden producir la síntesis de una proteína diferente o defectuosa y ocasionar una enfermedad. Que el individuo desarrolle o no una enfermedad a causa de la mutación dependerá de la importancia de la proteína en la función global del cuerpo humano. Si la mutación afecta el ADN de una célula germinal o reproductiva, se transmitirá a las siguientes generaciones y causará una enfermedad hereditaria.
Las enfermedades hereditarias se clasifican en:
1. Alteraciones cromosómicas, con la deleción o adición de una parte o un cromosoma entero.
2. Enfermedades poligénicas (las más frecuentes), en que la enfermedad se debe a la interacción de diferentes genes.
3. Enfermedades monogénicas. Sólo un gen es principalmente responsable de esta enfermedad, y su patrón de herencia sigue las leyes de Mendel, que son:
- Enfermedades autosómicas dominantes. Uno de los alelos heredados es defectuoso y el otro es normal. El carácter dominante de la enfermedad significa que con un solo alelo afectado por una mutación ya se puede provocar la enfermedad. La descendencia tiene un 50% de posibilidades de ser portadora de esta enfermedad si uno de los padres está afectado. Cada generación tiene afectados, y tanto los varones como las mujeres pueden heredar y transmitir la enfermedad.
- Enfermedades autosómicas recesivas. Para padecer la enfermedad se requiere que los dos alelos sean defectuosos. Es una forma, por tanto, menos común que la autosómica dominante. Si ambos padres son portadores, la descendencia tiene un 25% de posibilidades de padecer la enfermedad y un 50% de posibilidades de ser portadora.
- Enfermedades ligadas al sexo. Las mujeres son portadoras en uno de los cromosomas X. Los varones, al tener un solo cromosoma X, padecen la enfermedad si heredan el cromosoma mutado.
- Enfermedades mitocondriales. Están siempre transmitidas por la mujer, porque el cromosoma mitocondrial proviene siempre de la madre. Por tanto, no se produce transmisión de varón a varón.
En general, todos los seres humanos presentan pequeñas variaciones, llamadas polimorfismos, en un lugar determinado del ADN. A diferencia de las mutaciones, los polimorfismos no suelen causar enfermedades pero pueden alterar la respuesta del individuo a ellas, produciendo variaciones en la predisposición, la evolución y la respuesta al tratamiento8,9.
CONCEPTOS BÁSICOS: GENÉTICA, GENÓMICA, GENOTIPO Y FENOTIPO
La evolución que ha experimentado la biología molecular nos ha llevado, entre otros aspectos, al uso más cotidiano y a veces indiscriminado de un vocabulario específico. Es por ello que hemos considerado conveniente conceptualizar algunos de estos términos para poder enmarcar la temática que tratamos en este artículo.
En primer lugar, deberíamos diferenciar genética de genómica. Se entiende la genética como el campo de las ciencias biológicas que intenta comprender cómo la herencia biológica se transmite de una generación a la siguiente y cómo se desarrollan estos procesos. Así pues, uno de los objetivos de la genética es el estudio de los patrones de herencia y de cómo éstos se transmiten de padres a hijos. Por otra parte, la genómica incluye un conjunto de ciencias y técnicas dedicadas al estudio integral del funcionamiento, la evolución y el origen de los genomas6,10. A pesar de entender que genética y genómica son conceptualmente diferentes, tanto la genética como la genómica comparten unidades básicas de estudio: los genes y el ADN.
Genotipo y fenotipo son otros dos conceptos básicos que deben tenerse en cuenta para entender algunas cuestiones que se derivan de la investigación en este ámbito. El genotipo hace referencia a todo el material genético del individuo. El rasgo observable de un individuo se llama fenotipo6,10. Por ejemplo, al hablar de enfermedades, si un individuo puede tener una predisposición genética a padecer una enfermedad cardiovascular, como por ejemplo el síndrome de Brugada, nos estaríamos refiriendo al genotipo. Si esta enfermedad se manifiesta y se desarrolla, estamos hablando del fenotipo.
UNA CUESTIÓN DE DISEÑO
Charles Darwin, en su teoría del origen de las especies, pone de manifiesto que todo organismo tiene que esforzarse para sobrevivir, y que sólo el vigoroso, sano y feliz es capaz de multiplicarse. De hecho, puede decirse que la actividad física está programada en nuestros genomas desde la era paleolítica. Los factores ambientales con los que tuvieron que enfrentarse nuestros antepasados, donde la condición física era un factor muy importante para la supervivencia, ha hecho que nuestros genes hayan evolucionado en relación al ambiente en que vivieron nuestros ancestros11,12.
Los factores ambientales de las sociedades del bienestar son muy diferentes a los de la era del Paleolítico, por lo que nos encontramos con un diseño evolutivo de nuestro organismo incompatible con las condiciones medioambientales actuales. Este hecho provoca muchas de las enfermedades que hoy padece nuestra sociedad11.
Entre el genotipo y el fenotipo, los factores ambientales (alimentación, actividad física, hábitos higiénicos, etc.) desempeñan un papel muy importante. Sea de forma individual o colectiva, estos factores pueden interaccionar con el genotipo y ser precursores de la manifestación de fenotipos (fig. 1).
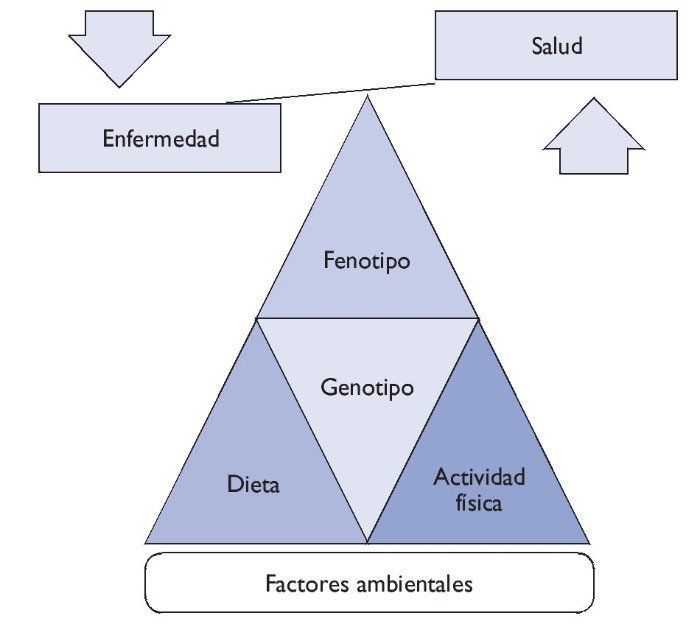
Figura 1 Relación genotipo-fenotipo.
GENOTIPOS Y FENOTIPOS DE LA CONDICIÓN FÍSICA
Hablar de condición física y genética nos lleva en primer lugar a determinar qué entendemos por condición física y cuáles son sus elementos. El concepto de condición física puede tener interpretaciones diferentes en función del ámbito en que nos encontremos. En relación a la temática de este artículo, queremos distinguir lo que es la condición física saludable y la condición física relacionada con el rendimiento deportivo.
En el ámbito de la salud, la condición física se puede entender como el grado de energía y vitalidad que consigue una persona para poder desarrollar las tareas diarias y habituales: disfrutar activamente del tiempo libre, afrontar las emergencias imprevistas sin fatiga excesiva, evitar algunas enfermedades derivadas del sedentarismo y ayudar a desarrollar al máximo su capacidad intelectual disfrutando plenamente de la vida. Desde el punto de vista del deporte, la condición física pasa a ser un elemento de rendimiento y se plantea como objetivo la máxima optimización de los componentes de la condición física13-16.
Con la finalidad de establecer un criterio que nos ayude a entender algunos aspectos de este artículo y sin querer entrar en debates conceptuales o de matices, consideramos que la condición física está formada por las capacidades físicas. Podemos clasificar estas capacidades físicas en capacidades motoras o básicas (fuerza, resistencia aeróbica/anaeróbica y velocidad), las capacidades coordinativas o perceptivo-motoras (coordinación general/específica y equilibrio), las capacidades resultantes (agilidad) y las capacidades facilitadoras (flexibilidad-elasticidad muscular/movilidad articular)14. Más allá de esta clasificación, estaríamos de acuerdo en que las capacidades condicionales se basan principalmente en procesos energéticos y las coordinativas preferentemente en la regulación y la conducción del sistema nervioso central15.
Sea desde la óptica deportiva o del ejercicio físico, todos estos elementos de la condición física pueden ser medidos de una forma o otra, desde el VO2máx (ml·kg-1·min-1 o l/min), el tiempo (s), la velocidad (m/s) o la fuerza (kg), etc. Así pues, y a modo de ejemplo, una persona puede tener un sustrato genético (genotipo) en relación a la resistencia cardiovascular que se puede manifestar en forma de valores de VO2máx (fenotipo) que es mesurable (ml·kg-1·min-1 o l/min) y modificable por las diferentes condiciones ambientales, como pueden ser las cargas de entrenamiento, la alimentación, etc.
Añadimos, con todo, que no debemos tener una visión mendeliana del individuo por lo que se refiere a los fenotipos de la condición física, ya que son muchos los factores que interaccionan: aspectos sociales, fisiológicos, metabólicos, celulares y moleculares. Además, los efectos del material genético sobre los fenotipos de la condición física también vienen determinados por los genes susceptibles que pueden predisponer a un fenotipo, y por las interacciones entre diferentes genes, cuestiones que hoy en día aún se están estudiando. Es por ello que los fenotipos de la condición física son fenotipos llamados complejos, como ocurre con los fenotipos de las enfermedades complejas (fig. 2).
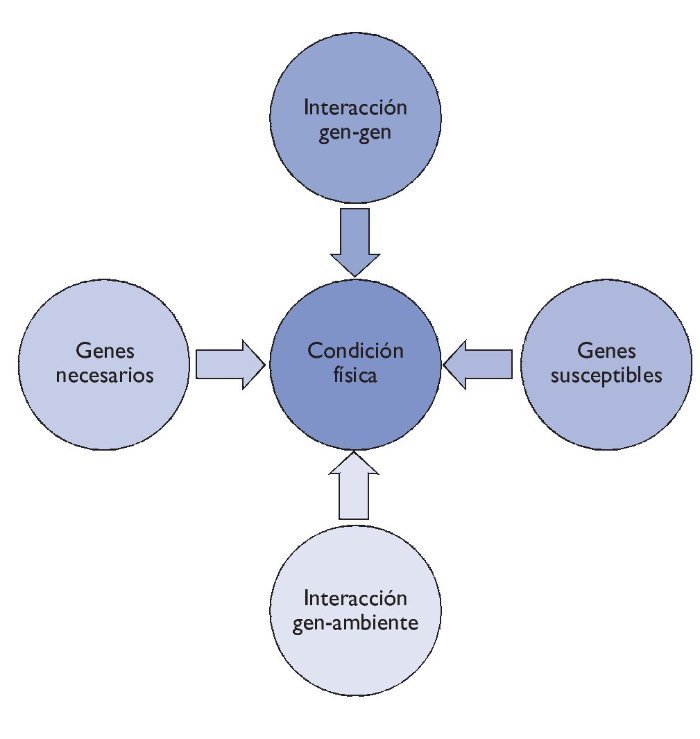
Figura 2 Interacción de los diferentes elementos en la condición física.
El mapa genético humano de la condición física
A partir de la finalización de la secuencia del genoma humano se han llevado y se llevan a cabo investigaciones para poder determinar genes implicados en los diferentes fenotipos de la condición física. Para elaborar este mapa genético se cuenta con las aportaciones de las diferentes investigaciones que se realizan año tras año. Con todo, la interpretación de los resultados de estos estudios ha de entenderse en relación a la estrategia utilizada en la investigación.
Estrategias de investigación
En estudios humanos los métodos de investigación se basan en la epidemiología genética y, más recientemente, en los estudios moleculares. A grandes rasgos, puede decirse que los estudios genéticos se pueden clasificar en dos grandes grupos: a) los basados en la epidemiología genética, centrados en fenotipos de individuos y familias, y b) los más relacionados con la biología molecular, que intentan medir las variaciones del ADN basándose en marcadores de genes candidatos.
Para analizar la base genética de fenotipos complejos, como pueden ser los de la capacidad física, hay tres grandes métodos17-20. En primer lugar, los estudios de heredabilidad, que intentan valorar la contribución genética y ambiental sobre los fenotipos de miembros de una misma familia, gemelos o individuos adoptados acogidos en familias. Por ejemplo, en algunos estudios realizados se estima que el grado de heredabilidad del VO2máx es aproximadamente del 50%; en cuanto al tipo de fibra muscular, de entre el 40 y el 50%, y la potencia muscular, alrededor del 70%.
Un segundo gran método es el Genome-wide linkage scans, que consiste en un examen genético de marcadores que se encuentran por todo el genoma humano en un grupo muy grande de individuos para posteriormente poder realizar asociaciones entre cada marcador y los fenotipos específicos. Se intentan localizar regiones del genoma humano que tengan una alta probabilidad de estar relacionadas con el consumo de oxígeno, el porcentaje de grasa corporal o la regulación de la frecuencia cardíaca.
La tercera gran estrategia y una de las más utilizadas son los estudios de asociación con gen candidato. Se trata de buscar un gen candidato que se cree que tiene una influencia en la regulación de alguno de los fenotipos de la condición física. A partir de aquí se intenta estudiar las variaciones más comunes de este gen (alelo) en un gran número de sujetos. En este bloque se pueden encontrar dos tipos de investigaciones: una estudia una única variación del gen (alelo) de unos sujetos en concreto, y la otra estudia la de los casos-control, donde se intenta ver si un gen o una variación de éste es más común en atletas de élite que en la población normal. Estas dos opciones de investigación son las que se han utilizado para identificar un número de genes asociados a la condición física, sea en la población en general o en atletas de élite.
Genes y condición física
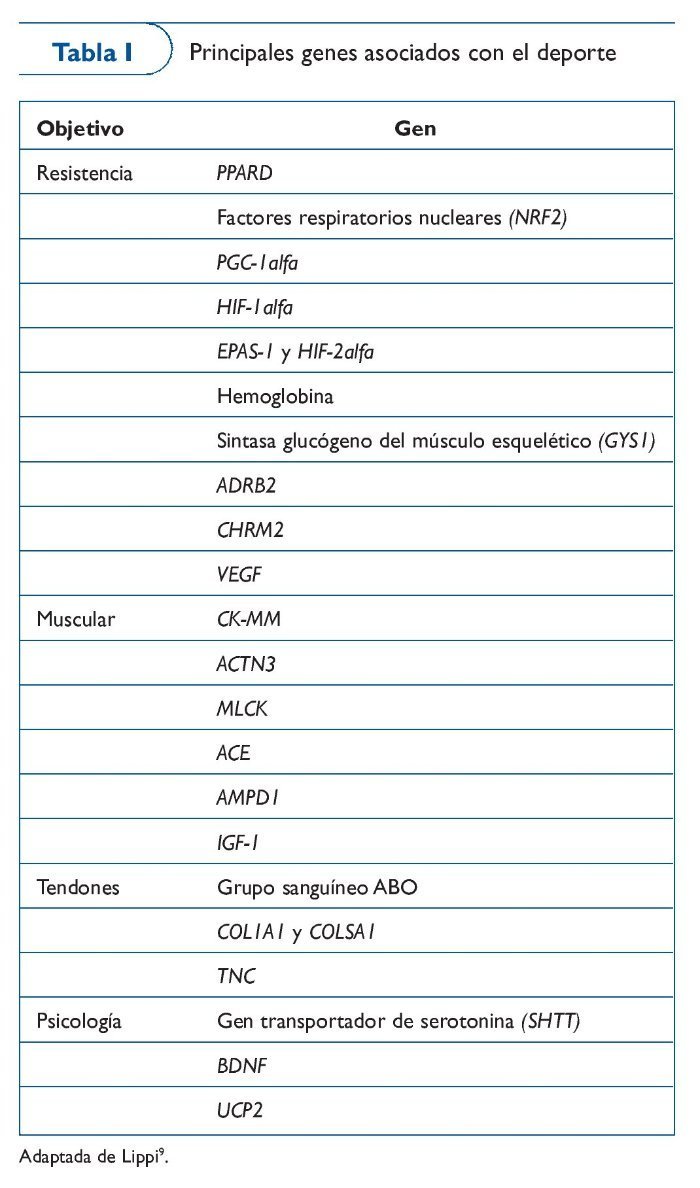
Los genes que se van añadiendo al genoma de la condición física aparecen publicados anualmente en The Human Gene Map for Performance and Health-Related Fitness Phenotypes. A pesar de que los estudios relacionados con los fenotipos de la condición física son muy variados, vamos a poner algunos ejemplos. En estudios de caso-control relacionados con modalidades deportivas de resistencia encontramos genes como AMPD1, PPARGC1A o ACE, y con pruebas de velocidad tenemos el ACTN3. Por lo que se refiere a estudios de asociación con genes candidatos encontramos fenotipos estudiados como el VO2máx asociado a genes como ADRB2, HLAA, CFTR o HIF1A, el lactato postejercicio asociado a genes como ACE, o la fuerza asociada a DI01 o IGF220 (tabla I).
Posiblemente las variantes genéticas tienen una incidencia tanto en el rendimiento deportivo como en las personas que practiquen ejercicio físico con el objetivo de mejorar la salud. En general, estas variantes estudiadas en deportistas de élite son muy comunes en la población. Los genes relacionados con el metabolismo energético, la respuesta cardiorrespiratoria al ejercicio máximo, etc., son aspectos importantes tanto para la población en general como para los deportistas. Si bien el estudio de las características genéticas de atletas de alto nivel aporta una información muy valiosa sobre los beneficios del ejercicio físico en la población general, también cabe decir que no todas las variantes que pueden encontrarse en deportistas de primera línea pueden ser un referente de salud en el mundo del fitness. Éste sería el caso de las variantes genéticas que puede tener un corredor de larga distancia y que le permiten conservar energía durante largos períodos de tiempo de intensa actividad física, ya que esto no podría ser beneficioso en individuos poco activos o sedentarios.
GENÉTICA Y MUERTE SÚBITA EN EL ATLETA
La muerte súbita cardíaca (MSC) se define como la muerte de causa cardíaca que se produce en la primera hora después de los primeros síntomas. La muerte súbita tiene una prevalencia de 1/1.000 en la población general, pero aumenta significativamente a medida que hay una patología cardíaca más grave. La muerte súbita en los atletas parece ser poco frecuente21,22, a pesar de que la verdadera incidencia puede ser más grande de lo que se cree debido a que ciertas condiciones -como las canalopatías iónicas, que predisponen a las arritmias mortales- no se asocian con enfermedades estructurales del corazón y esto supone que la causa de la muerte siga sin estar clara. Aproximadamente el 80% de las muertes súbitas no traumáticas en atletas jóvenes se deben a anomalías cardiovasculares heredadas, que por tanto tienen una repercusión en las familias23-25. La mayoría (>80%) de las MSC en los jóvenes atletas se producen durante o inmediatamente después de la actividad física26. Esto sugiere que el ejercicio puede ser un desencadenante de las arritmias cardíacas en individuos con ciertos trastornos cardíacos.
La mayoría de las MSC de los atletas se dan en varones jóvenes (12-40 años). A menudo la MSC es el primer síntoma de la enfermedad. La MSC suele estar directamente relacionada con la práctica de un ejercicio, y al realizar la autopsia ésta puede ser completamente normal (sospechosa de enfermedad arritmogénica) o detectarse alteraciones estructurales cardíacas24,26-28. Entre las enfermedades estructurales se han descrito sobre todo la cardiomiopatía hipertrófica (CMH), que causa del 40 al 50% de los casos, y la displasia arritmogénica del ventrículo derecho (DAVD), la causa más frecuente de MSC en atletas en ciertas zonas de Italia.
Existen, pues, múltiples patologías cardíacas genéticamente determinadas, con o sin cardiopatía estructural acompañante, que pueden predisponer a la aparición de arritmias y de muerte súbita29-31. Estas enfermedades son producto de la alteración en la codificación genética de cuatro grandes familias de proteínas:
Proteínas sarcoméricas, que se encargan de generar la fuerza en el miocito cardíaco y son responsables de la miocardiopatía hipertrófica32.
Proteínas del citoesqueleto, encargadas de la transmisión de esta fuerza a las células adyacentes y que causan la miocardiopatía dilatada33.
Proteínas que codifican por los canales iónicos, encargadas de mantener el equilibrio iónico intra y extracelular, y responsables de las arritmias familiares34.
Proteínas de los desmosomas, que permiten el mantenimiento de la estructura y la comunicación intercelular.
Con todo, se da un importante solapamiento entre las enfermedades y los genes35. Por ejemplo, la troponina T puede ser causante tanto de miocardiopatía dilatada como de miocardiopatía hipertrófica36,37, y el canal de sodio SCN5A es uno de los genes responsables del síndrome de Brugada (SBr), del síndrome de QT largo tipo 3 (SQTL3) y también de alteraciones familiares de la conducción38,39.
ENFERMEDADES
Síndrome de Brugada
El SBr tiene una elevación característica del segmento ST que es fácilmente identificable en el "patrón clásico" de los casos. Desgraciadamente son comunes en la población general los cambios no específicos del segmento ST o cambios incompletos de bloqueo de rama derecha. Ante la sospecha de la enfermedad, en estos casos menos clásicos se aconseja la prueba de la ajmalina, un bloqueador del sodio que, administrado por vía intravenosa, puede hacer evidente el patrón electrocardiográfico diagnóstico.
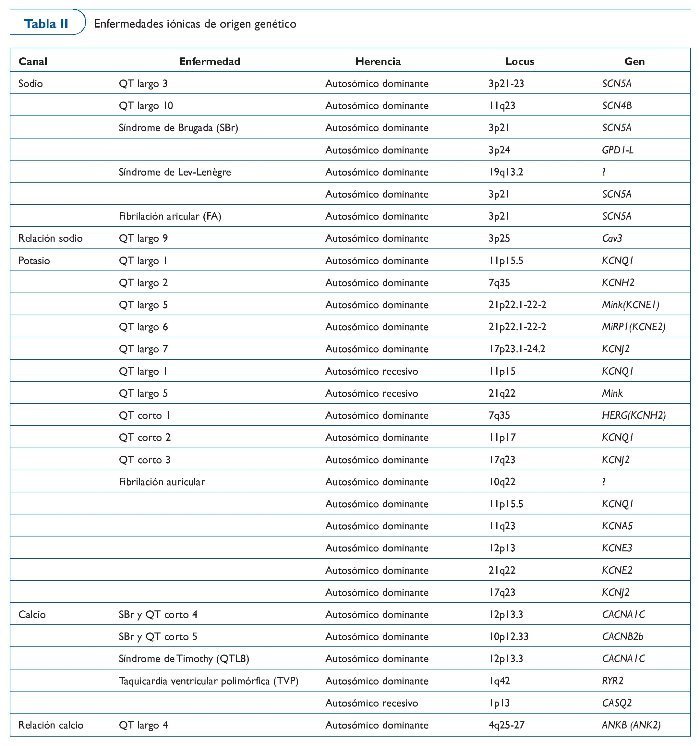
Se cree que el mecanismo fisiopatológico responsable de la elevación del ST y de la susceptibilidad de arritmias ventriculares es el desequilibrio entre entradas y salidas de corrientes iónicas durante la fase 0 y 1 del potencial de acción. Hasta ahora, sólo dos genes se han relacionado con la enfermedad (SCN5A, codificación de los canales de sodio, y GPD1L, el glicerol-3-fosfato deshidrogenasa-1). Curiosamente, no hay información respecto la muerte súbita en atletas causada por SBr, pero la práctica clínica ha aconsejado últimamente que los afectados por la enfermedad no practiquen deporte debido al aumento de la temperatura corporal, una decisión como mínimo controvertida (tabla II).
Taquicardia ventricular polimórfica catecolaminérgica
La taquicardia ventricular polimórfica catecolaminérgica (TVPC) es una enfermedad familiar que causa MSC inducida por el ejercicio y/o el estrés. La TVPC es un trastorno del calcio intracardíaco agravado por la estimulación simpática. Mutaciones en el receptor de rianodina (Ryr2), calsiquestrina-2 (CASQ2) y ankirina-2 (ANK2) producen una sobrecarga de calcio que se ha relacionado con la enfermedad (tabla II).
Síndrome del Qt largo
El síndrome del QT largo (SQTL) se caracteriza por la prolongación del intervalo QT en el ECG de reposo (> 470 ms para los varones y > 480 ms para las mujeres como diagnóstico, y de 440-470 ms como límite). El síndrome estuvo inicialmente relacionado con la pérdida de función en los canales de potasio y el incremento de la función en el canal de sodio. En todos los casos, la consecuencia es un potencial de acción prolongado que facilita despolarizaciones, un sustrato de arritmias ventriculares (tabla II).
Clínicamente, hasta el 30% de los casos de SQTL tienen un intervalo QT en el límite de la normalidad que requiere otros estudios (fenotípicos, genéticos o ambos) para llegar a un diagnóstico. Dada la variedad de mecanismos que conducen a la SQTL, la identificación de las mutaciones causales es crucial para orientar la terapia. No obstante, los análisis genéticos son negativos en un tercio de los pacientes, lo que acentúa la necesidad de mejorar la detección fenotípica.
Síndrome del Qt corto
El síndrome del QT corto (SQTC) se caracteriza por la presencia de acortamiento del QT en el ECG (QT < 340 ms es sospechoso, y < 320 es claramente diagnóstico) y clínicamente por la presencia de episodios de síncope, fibrilación auricular paroxística y/o arritmias letales. A pesar de que se han publicado algunos casos de familias afectadas, se dispone de poca información sobre la enfermedad. El SQTC se ha relacionado con mutaciones en los genes que codifican canales de potasio. Hasta ahora se han identificado tres genes, y este hecho demuestra que la enfermedad es genéticamente heterogénea. A pesar de que los estudios clínicos y fisiopatológicos han sugerido la quinidina como un tratamiento adecuado para STQC-1, la alta incidencia de MSC justifica la implantación de desfibrilador en la mayoría de los casos (tabla II).
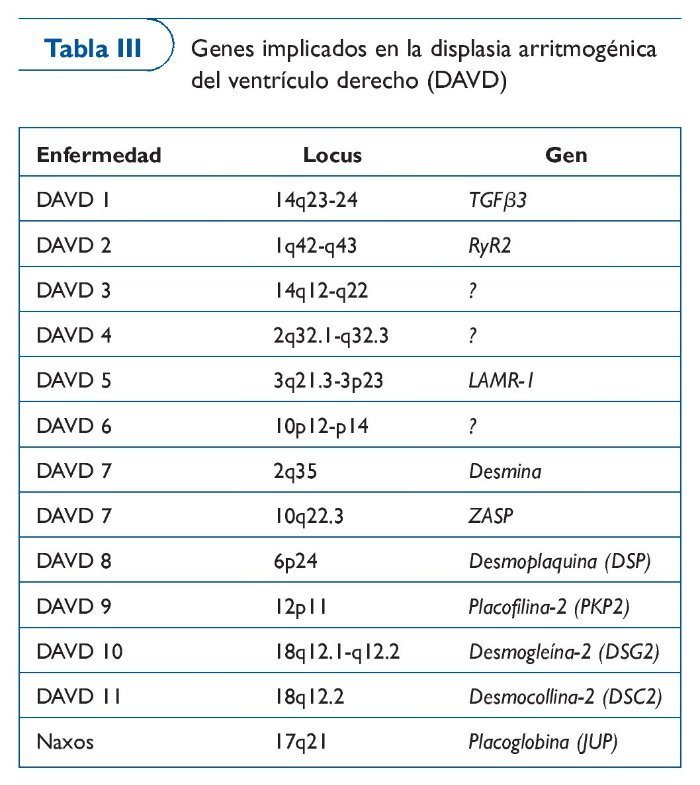
Displasia arritmogénica de ventrículo derecho
La DAVD es una enfermedad familiar40 del músculo cardíaco que provoca cambios estructurales (distrofia progresiva del ventrículo derecho, con sustitución del miocardio por tejido fibroadiposo) y clínicamente se caracteriza por arritmias del ventrículo derecho, con riesgo de paro cardíaco41,42.
A pesar de que en los últimos 25 años se ha descrito la enfermedad43, ciertas formas de ARVC no son fáciles de diagnosticar. El diagnóstico precoz de la enfermedad es difícil debido a que no hay un patrón claro de afectación de los tejidos. La resonancia magnética es una herramienta muy útil en el diagnóstico, pero la interpretación es difícil en algunos casos. Además, en algunos pacientes la primera manifestación son las arritmias ventriculares malignas. Las pruebas genéticas ayudan al diagnóstico clínico, dado que ya se puede diagnosticar la enfermedad en cerca del 60% de los casos gracias a los estudios genéticos, encaminados al análisis de mutaciones en proteínas del desmosoma (tabla III).
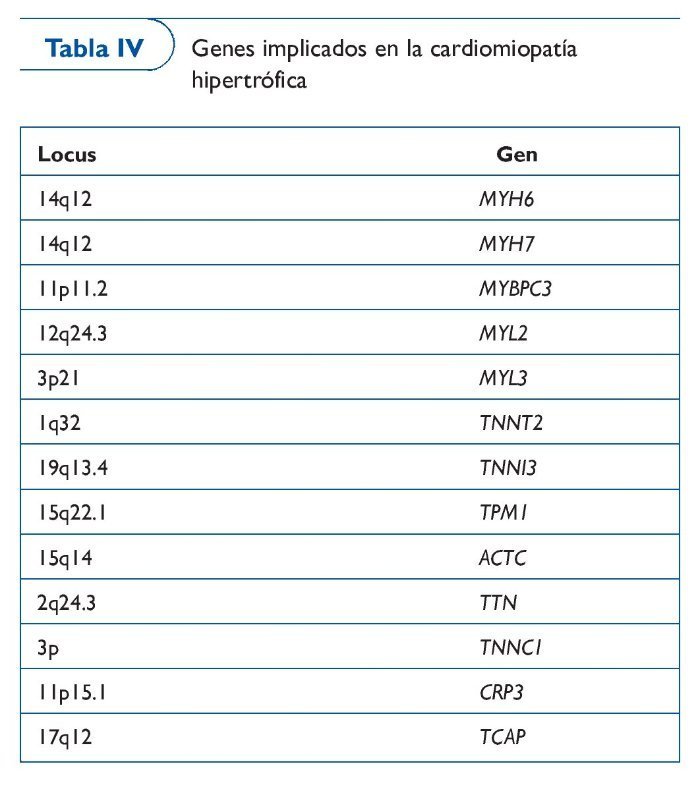
Cardiomiopatía hipertrófica
La CMH es una enfermedad del miocardio caracterizada por una hipertrofia asimétrica del ventrículo izquierdo, con hallazgos de desestructuración de los miocitos y fibrosis44,45. Es la alteración genética cardiovascular más frecuente, con una prevalencia de 1/500 en la población general, y afecta sobre todo a jóvenes46-48. Las manifestaciones clínicas aparecen por disfunción diastólica inicialmente y sistólica en los estados más evolucionados, por lo que el paciente puede estar asintomático o presentarse en forma de insuficiencia cardíaca o muerte súbita. La mortalidad es mayor en los pacientes jóvenes (a menudo atletas) que en los adultos, y la primera manifestación de la enfermedad puede ser precisamente la muerte súbita. La enfermedad se considera familiar en un 90% de los casos, generalmente con un patrón de herencia autosómica dominante, a excepción de los casos con mutaciones en el ADN mitocondrial (ADNmt), que son heredadas por vía materna. Se han identificado más de 400 mutaciones49-50 (tabla IV).
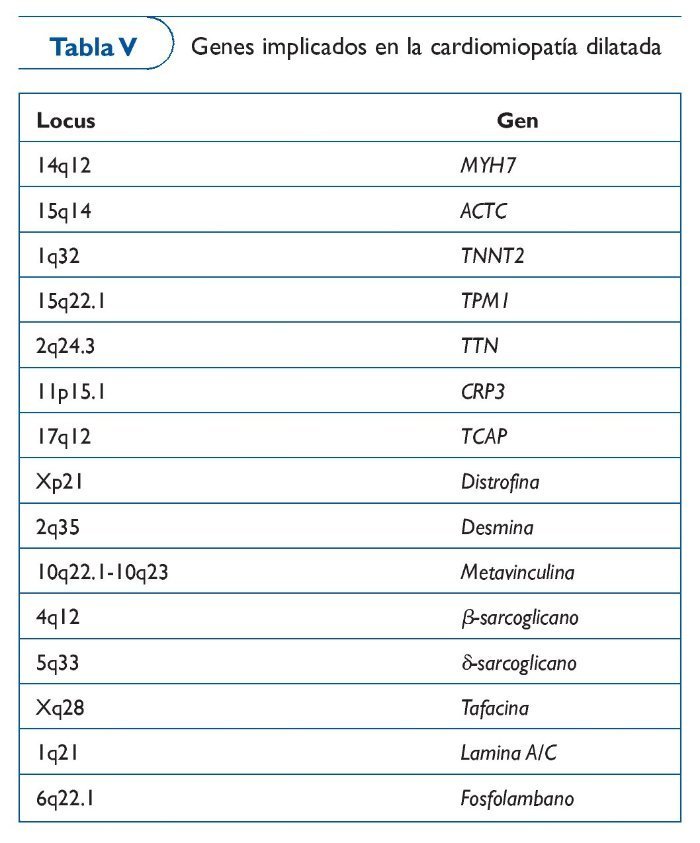
Cardiomiopatía dilatada
La cardiomiopatía dilatada (CMD) se caracteriza por dilatación ventricular que altera la función sistólica, principalmente en el ventrículo izquierdo. Los pacientes presentan signos de insuficiencia cardíaca, palpitaciones o muerte súbita. La prevalencia es de 1/2.500 personas. Son múltiples los factores que pueden desencadenar una CMD, por lo que es una entidad altamente heterogénea. A pesar de ello, los estudios sistemáticos de los familiares de pacientes con CMD indican que al menos el 35% de los casos son hereditarios51. Las arritmias que presentan los pacientes con CMD familiar suelen ser las mismas que en las formas adquiridas con defectos en la conducción auriculoventricular e intraventricular, arritmias ventriculares y fibrilación auricular. Estos pacientes normalmente desarrollan un deterioro progresivo de la función ventricular y mueren por insuficiencia cardíaca o arritmias.
En 1994 de encontró el primer locus de CMD con bloqueo auriculoventricular en el cromosoma 152. El escenario de esta enfermedad es extremadamente complejo, por lo que la utilidad clínica de análisis genético es limitada. Se han identificado más de 20 mutaciones en genes que codifican por proteínas del citoesqueleto, del núcleo celular y del sarcómero. Las mutaciones más importantes (30%) son las encontradas en el gen lámina A/C (LMNA)53, que codifica una proteína que se expresa en casi todos los tipos celulares y cuya función es contribuir con la integridad del núcleo proporcionando soporte mecánico54. Otros genes, como MYH7 y TNNT2, identificados previamente como causantes de CMH, también pueden causar CMD. Incluso en una familia con CMD se identificó mutación en SCN5A, un gen que se creía únicamente causante de enfermedad eléctrica55 (tabla V).
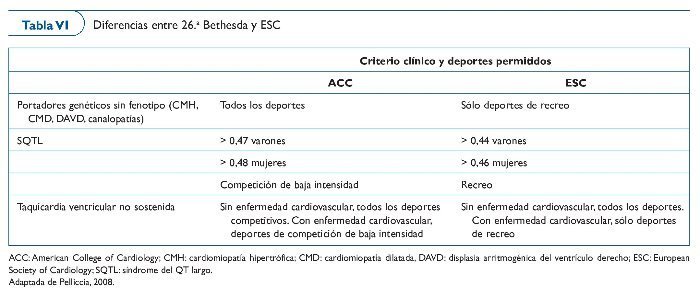
RECOMENDACIONES DEPORTIVAS EN LAS ENFERMEDADES GENÉTICAS
El 1994 se organizó la 26.a Conferencia Bethesda (tabla VI) para formular las directrices de la participación en deportes competitivos de atletas con alguna alteración cardiovascular identificada56. Expertos en medicina cardiovascular y cardiología del deporte redactaron recomendaciones por medio de un consenso, proporcionando la base para el asesoramiento médico a los pacientes. Las directrices dependen de la naturaleza y de la gravedad de las anomalías cardiovasculares y de la clasificación del deporte en cuestión.
Casi en otra tercera parte de las MSC la autopsia no revela ninguna anomalía estructural o morfológica. Se admite que en estos casos el origen de la muerte podría ser puramente eléctrico y, en el caso de enfermedad familiar, una canalopatía. Entidades conocidas, como la taquicardia ventricular polimórfica, la fibrilación ventricular idiopática, el SQTL, el SQTC y el SBr, son entidades englobadas bajo este epígrafe de canalopatías.
La muerte súbita de un joven atleta tiene habitualmente una repercusión mediática importante. Debido al sobreesfuerzo que tienen que desarrollar, hay criterios claros sobre su participación en deportes de competición56-60.
Atletas con un diagnóstico claro de CMH, de DAVD y de CMD no deben participar en la mayoría de deportes de competición, con la posible excepción de los deportes con el menor grado de intensidad (p. ej., bolos, golf o curling). Esto es independiente de la presencia de síntomas y de la magnitud de la hipertrofia ventricular izquierda del ventrículo izquierdo o la obstrucción del tracto de salida en el caso de la CMH.
Se recomienda no practicar deporte de competición en las enfermedades genéticas arritmogénicas, sobre todo por las implicaciones que el estado adrenérgico puede tener en el desencadenamiento de arritmias. La recomendación en el SBr es menos clara, pero debido a la asociación de la enfermedad con la fiebre, se ha considerado necesario también desaconsejar la práctica del deporte por el aumento de temperatura corporal que esto comporta.
VALORACIÓN DE LOS ATLETAS CON RASGOS CARACTERÍSTICOS DE POSIBLE ENFERMEDAD CARDÍACA
La valoración cardiovascular en el atleta joven se ha dirigido a identificar las condiciones que pueden llevar al atleta al riesgo de muerte súbita. Idealmente, todos los atletas tendrían que ser evaluados, como mínimo, para diagnosticar posibles alteraciones cardiovasculares antes de cualquier participación atlética, pero quienes practican deporte regularmente deberían disponer de una valoración clínica básica (examen físico e historia clínica) y de un electrocardiograma en reposo.
Tras las exploraciones clínicas básicas en los atletas, es importante someter a estudios más profundos a los que tengan mayor riesgo de presentar anomalías cardíacas hereditarias. Éstos son, por supuesto, los atletas sintomáticos y los que tienen historia familiar de enfermedad familiar asociada a la muerte súbita.
Entre los estudios para realizar se puede ya actualmente considerar el estudio genético, que es efectivo en los casos de enfermedad hereditaria conocida, pero lo es poco si la causa de la muerte súbita o de los síntomas todavía no es conocida. En este último caso, la cantidad de genes que hay que analizar es tan grande que no es asumible económicamente. En los próximos meses incluso estos casos podrán ser analizados rápidamente, gracias al desarrollo tecnológico de chips de diagnóstico de enfermedades arrítmicas. Este chip permitirá realizar el análisis de más de 20 genes en un margen reducido de tiempo, mejorando el servicio y, sobre todo, el coste.
CONCLUSIONES
Al largo de la historia, la sociedad ha encontrado un sitio especial para los pocos que son más rápidos, más fuertes y físicamente mejor dotados. En la actualidad empieza a haber estudios lo suficientemente consistentes que indican que los genes pueden desempeñar un papel importante en la condición física de los deportistas de élite, a pesar de que aún es pronto para determinar si un único gen o grupo de genes puede determinar el potencial deportivo de un sujeto. La genética puede determinar también el riesgo de muerte súbita en el atleta. Cuando se produce una MSC en un deportista, es una noticia trágica y con mucho impacto social.
Así pues, es evidente que en las próximas décadas se abre un campo de investigación al que tendremos que incorporarnos, y habremos de adaptarnos a tecnologías de la genómica y la bioinformática con el objetivo de conocer la influencia de la genética en la condición física de los individuos y en su riesgo de enfermedades.
Correspondencia: Ramon Brugada (ramon@brugada.org).
Recibido el 8 de mayo de 2009 / Aceptado el 13 de mayo de 2009

